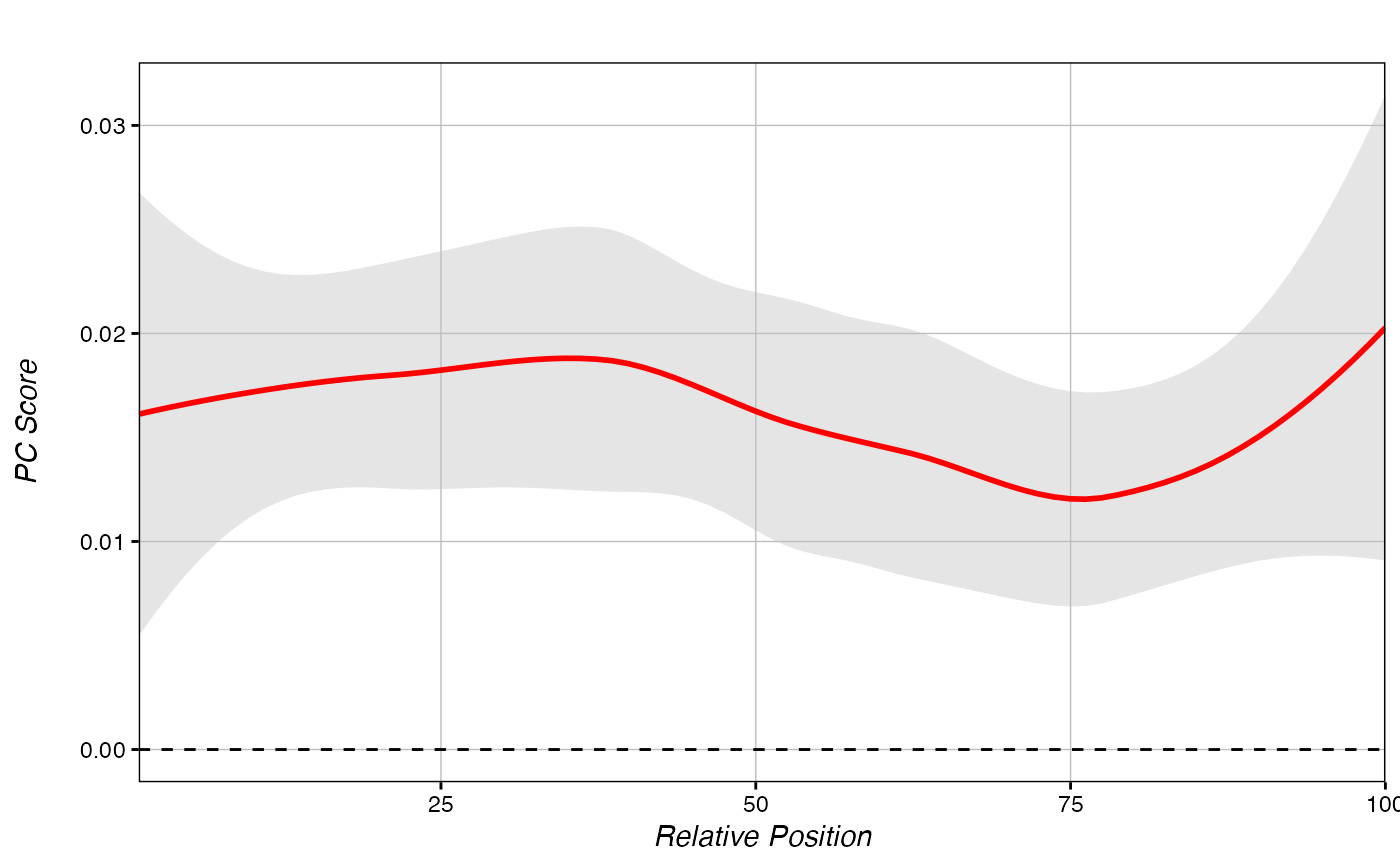
Make a metagene from PC Scores.
score_metagene.RdUses mean binned PC scores across a set of regions to draw a metagene.
score_metagene(ranks, regions, bin, title, xaxis, yaxis,
chromDictObj, return.data, linecol)Arguments
- ranks
getPCRanks output data.frame
- regions
A three-column data.frame containing a set of regions to test. Columns = chrom, start, end.
- bin
integer, number of bins to use in metagenes. Default=100.
- title
Output plot title
- xaxis
Output plot x-axis title
- yaxis
Output plot y-axis title
- chromDictObj
Optional chromDictObject made from chromDict(), runs internally if set to NULL (default). Scripts that run this function multiple times will be sped up by setting this option.
- return.data
T/F, whether to return a plot, or data that can be run with plot_metagene() or multiple_metagenes().
- linecol
Colour for line, auto="red"
Value
If return.data=F, returns a grob containing a metagene plot. Otherwise, returns a list of two data.frames containing metagene and metagene standard error plotting information.
Examples
ranks <- getPCRanks(eigen, IDs = c("trt", "ctl"), PC = 1)
DMRs <- Get_Novel_DMRs(ranks, 2940, minCpGs=10)
#> Splitting data by chromosome...
#> Bootstrapping background distributions for each chromosome...
#> Compressing nearby seeds...
#> done! Collapsed 2940 seeds to 250 seeds!
#>
#> Expanding DMRs from 250 seeds...
#> Trimming 135 DMRs...
#> done!
# select chrom, start, and end of all hyper DMRs:
hyper_DMRs <- DMRs[DMRs$FDR <= 0.05 & DMRs$DMR_Zscore > 0,]
regions_hyper <- hyper_DMRs[c(1:3)]
score_metagene(ranks, regions_hyper)
#> Creating chromDict Object. If you plant to run the score_metagene() function multiple times, it is strongly recommended to generate a chromDict Object with the chromDict() function, and specify it with score_metagene(..., chromDictObj=OBJECT). This is the most computationally intensive part of metagene creation, and only needs to be done once.